Steppe Ancestry in western Eurasia and the spread of the Germanic Languages (McColl et al.) The Genetic Origin of the Indo-Europeans (Lazaridis et al.) A genomic history of the North Pontic Region from the Neolithic to the Bronze Age (Nikitin et al.)All of these studies are very useful, but there are some problems with each of them. Indeed, I'd say that the authors of the Lazaridis and McColl preprints need to reevaluate the way that they use ancient DNA to solve their linguistic puzzles. Once they do that their conclusions are likely to change significantly. I'm aiming to produce a couple of detailed blog posts about these preprints within the next few weeks. Afterwards I'll get in touch with the relevant authors to change their minds about some key things. Please stay tuned. See also... Indo-European crackpottery
Friday, April 19, 2024
It's complicated
Three important manuscripts appeared recently at bioRxiv, mostly dealing with the origins and expansions of proto-Germanic and proto-Indo-European populations.
Monday, March 25, 2024
High-resolution stuff
I just emailed this to the authors of High-resolution genomic ancestry reveals mobility in early medieval Europe, a new preprint at bioRxiv [LINK].
I appreciate that Polish population history is not the main focus of your preprint, and also that you're constrained by the lack of relevant and suitably high quality ancient genomes from East-Central and Eastern Europe. However, I must say that your analysis of the Medieval Polish population and resulting conclusions about Polish population history don't reflect reality. Your Poland_Middle_Ages genomic cluster is made up of just six samples that don't fully represent the genetic complexity of the core population of Medieval Poland. As a result, you classified PCA0148 as one of the Poland_Middle_Ages outliers, even though this sample isn't an outlier when analyzed within the context of the full set of published Polish Medieval genomes. Moreover, PCA0148 is very similar to several Polish Viking Age samples that show Scandinavian-specific genome-wide and Y-chromosome haplotypes, and probably likewise shows some Scandinavian-related ancestry. This is important to note when attempting to recapitulate Polish population history, because it suggests that Scandinavian-related ancestry played a formative role in the shaping of the core Polish Medieval genetic cluster. Thus, you might be correct when you claim that the six samples in your Poland_Middle_Ages cluster don't show any "detectable" Scandinavian-related ancestry, but this doesn't necessarily mean that this type of ancestry isn't a key part of the post-Iron Age Polish population history. Below is a self-explanatory Principal Component Analysis (PCA) plot that illustrates my points. Interestingly, Figure 3c in your preprint shows very similar outcomes in regards to the post-Iron Age Polish population history. But the style and scale of your figure makes it difficult to spot the subtle but likely genuine Northwest European-related genetic shifts shown by PCA0148, the Viking context samples and present-day Poles relative to the Poland_Middle_Ages cluster.Update 26/03/2024: I sent another email to Speidel et al., this time in regards to their analysis of present-day Hungarians.However, I'm also skeptical that your Poland_Middle_Ages cluster doesn't carry any detectable or even significant Scandinavian-related ancestry. That's because I suspect that there might be some technical issues with your analysis that are masking this type of ancestry in the Polish samples. Your top mixture model for the Poland_Middle_Ages cluster is, in all likelihood, an extreme statistical abstraction of reality, rather than a close reflection of it. That's because, due to a combination of historical, geographical and genetic factors, neither Italy.Imperial(I).SG nor Lithuania.IronRoman.SG are realistic formative source populations for the Medieval Polish gene pool. One of the reasons why you ended up with such a surprising result is probably the lack of suitable samples from East-Central and Eastern Europe, especially those associated with plausibly the earliest Slavic-speaking populations. It's also possible that basing your mixture model on formal statistics played a key part. Formal statistics-based mixture models are known to be biased towards outcomes involving mixture sources from the extremes of mixture clines. If your analysis is affected by this problem, then this would help to explain why you characterized the Poland_Middle_Ages cluster as simply a two-way mixture between a Middle Eastern-related group from Imperial Rome and a Baltic population with a very high cut of European hunter-gatherer ancestry. I do note that on page 6 of your manuscript you consider the possibility that the Southern European-related signal in the Poland_Middle_Ages cluster might only be very distantly related to Italy.Imperial(I).SG, and that it may even have spread across Poland with early Slavic speakers. This is a great point, and I think it should be emphasized and expanded upon, because I suspect that the problem runs deeper than this. For instance, if the early Slavic ancestors of Poles carried substantially more Southern European-related ancestry than Lithuania.IronRoman.SG, and this ancestry was, say, more Balkan-related than Italian-related, then this might radically change your modeling of the Poland_Middle_Ages cluster. That's because these early Slavs would be positioned in a very different genetic space than Lithuania.IronRoman.SG, which could potentially require a significant signal of Scandinavian-related ancestry to get a robust mixture model. Finally, it might be useful to consider Isolation-by-Distance as a partial vector for the Italy.Imperial(I).SG-related signal in Medieval Poland. The full set of published Polish Medieval genomes includes a number of outliers with obvious ancestry from Western Europe and the Balkans. These people probably don't represent any large-scale migrations into Poland, but rather the movements of individuals and small groups. Over time, such small-scale mobility may have had a fairly significant impact on the genetic character of the Polish population.

Your preprint also claims that present-day Hungarians are genetically similar to Scythians, and that this is consistent with the arrival of Magyars, Avars and other eastern groups in this part of Europe. However, present-day Hungarians are overwhelmingly derived from Slavic and German peasants from near Hungary. This is not a controversial claim on my part; it's backed up by historical sources and a wide range of genetic analyses. Hungarians still show some minor ancestry from Hungarian Conquerors (early Magyars), but this signal only reliably shows up in large surveys of Y-chromosome samples. The Scythians that you used to model the ancestry of present-day Hungarians are of local, Pannonian origin, and they don't show any eastern nomad ancestry. So they're either acculturated Scythians, or, more likely, wrongly classified as Scythians by archeologists. And since these so-called Scythians lack eastern nomad ancestry, the similarity between them and present-day Hungarians is not a sign of the impact from Avars, Hungarian Conquerors and the like, but rather a lack of significant input from such groups in present-day Hungarians.Citation... Speidel et al., High-resolution genomic ancestry reveals mobility in early medieval Europe, bioRxiv, Posted March 19, 2024, doi: https://doi.org/10.1101/2024.03.15.585102 See also... Wielbark Goths were overwhelmingly of Scandinavian origin
Thursday, February 22, 2024
Berkeley, we have a problem
A new preprint at bioRxiv by Kerdoncuff et al. makes the following, somewhat surprising, claim:
 Citation...
Kerdoncuff et al., 50,000 years of Evolutionary History of India: Insights from ∼2,700 Whole Genome Sequences, bioRxiv, posted February 20, 2024, doi: https://doi.org/10.1101/2024.02.15.580575
See also...
The Nalchik surprise
A comedy of errors
Citation...
Kerdoncuff et al., 50,000 years of Evolutionary History of India: Insights from ∼2,700 Whole Genome Sequences, bioRxiv, posted February 20, 2024, doi: https://doi.org/10.1101/2024.02.15.580575
See also...
The Nalchik surprise
A comedy of errors
One of the individuals, referred to Sarazm_EN_1 (I4290) described above that was discovered with shell bangles showing affiliation with South Asia, has significant amount AHG-related ancestry, while a model without AHG-related ancestry provides the best fit for Sarazm_EN_2 (I4210) (Table S4.5).First of all, the authors are actually referring to sample ID I4910 not I4210. The aforementioned table, based on qpAdm output, shows that I4290 has 15.9% AHG-related ancestry and basically no Anatolian farmer-related ancestry. It also shows that I4910 has no AHG-related ancestry but 17.9% Anatolian farmer-related ancestry. AHG stands for Andaman hunter-gatherer. The authors are using it as a proxy for South Asian hunter-gatherer ancestry. However, I've looked at I4290 and I4910 in great detail over the years using ADMIXTURE, Principal Component Analysis (PCA), and qpAdm. And I'm quite certain that they do not show any obvious, above noise level South Asian ancestry. Indeed, I'd say that if they do have some minor South Asian ancestry, then I4910 probably has more of it than I4290. Kerdoncuff et al. used the following "right pops" or outgroups: Ethiopia_4500BP.SG, WEHG, EEHG, ESHG, Dai.DG, Russia_Ust_Ishim_HG.DG, Iran_Mesolithic_BeltCave and Israel_Natufian. This means they mixed data that were generated in very different ways (DG, SG and capture) and included some poor quality samples. For instance, the highest coverage version of Iran_Mesolithic_BeltCave offers just ~50K SNPs. Mixing different types of data and relying on low coverage samples, even in part, often has negative consequences when using qpAdm. So I suspect that the above mentioned mixture results for I4290 are skewed by a poor choice of outgroups. When I run qpAdm I try to stick to one type of data and avoid low quality singletons in the outgroups. This is the best qpAdm model that I can find for Sarazm_EN:
right pops: Cameroon_SMA Morocco_Iberomaurusian Israel_Natufian Levant_N Iran_GanjDareh_N Turkey_N Russia_Karelia_HG Russia_WestSiberia_HG Mongolia_North_N Brazil_LapaDoSanto_9600BP Sarazm_EN Kazakhstan_Botai_Eneolithic 0.113±0.017 Turkmenistan_C_Geoksyur_subset 0.887±0.017 P-value 0.06392 Sarazm_EN_1 (I4290) Kazakhstan_Botai_Eneolithic 0.129±0.021 Turkmenistan_C_Geoksyur_subset 0.871±0.021 P-value 0.11019 Sarazm_EN_2 (I4910) Kazakhstan_Botai_Eneolithic 0.104±0.021 Turkmenistan_C_Geoksyur_subset 0.896±0.021 P-value 0.07427 Also... Sarazm_EN Andaman_hunter-gatherer -0.018±0.020 Kazakhstan_Botai_Eneolithic 0.123±0.019 Turkmenistan_C_Geoksyur_subset 0.895±0.020 P-value 0.0298403 (Infeasible model)Please note that Turkmenistan_C_Geoksyur_subset is made up of just three relatively high quality individuals: I8504, I12483 and I12487. That's because it's not possible to model the ancestry of Sarazm_EN using the full Geoksyur set, probably due to subtle genetic substructures within the latter. Below is a PCA plot that, more or less, reflects my qpAdm model. I4290 and I4910 are sitting right next to each other in a cluster of ancient Central and Western Asians, and it's actually I4910 that is shifted slightly towards the South Asian pole of the PCA. Indeed, I can confidently say that there's no way to design a PCA in which I4290 is shifted significantly towards South Asia relative to I4910.

Monday, February 12, 2024
The Nalchik surprise
If, like Iosif Lazaridis, you subscribe to the idea that the Yamnaya people carry early Anatolian farmer-related admixture that spread into Eastern Europe via the Caucasus, then I've got great news for you.
We now have a human sample from the Eneolithic site of Nalchik in the North Caucasus, labeled NL122, that packs well over a quarter of this type of ancestry (see here). Below is a quick G25/Vahaduo model to illustrate the point (please note that Turkey_N = early Anatolian farmers).
 Indeed, I also don't exactly understand the recent infatuation among many academics, especially Iosif Lazaridis and his colleagues, with trying to put the Proto-Indo-Anatolian homeland somewhere south of the Caucasus. Considering all of the available multidisciplinary data, I'd say it still makes perfect sense to put it in the Sredny Stog culture of the North Pontic steppe, in what is now Ukraine.
Please note that all of the G25 coordinates used in my models and the PCA are available HERE.
See also...
The Caucasus is a semipermeable barrier to gene flow
Indeed, I also don't exactly understand the recent infatuation among many academics, especially Iosif Lazaridis and his colleagues, with trying to put the Proto-Indo-Anatolian homeland somewhere south of the Caucasus. Considering all of the available multidisciplinary data, I'd say it still makes perfect sense to put it in the Sredny Stog culture of the North Pontic steppe, in what is now Ukraine.
Please note that all of the G25 coordinates used in my models and the PCA are available HERE.
See also...
The Caucasus is a semipermeable barrier to gene flow
Target: Nalchik_Eneolithic:NL122 Distance: 2.1934% / 0.02193447 60.8 Russia_Steppe_Eneolithic 26.2 Turkey_N 13.0 Georgia_KotiasOn the other hand, if, again like Iosif Lazaridis, you subscribe to the idea that the Indo-European language spread into Eastern Europe via the Caucasus in association with this early Anatolian farmer-related admixture, then I've got terrible news for you. That's because NL122 is apparently dated to a whopping 5197-4850 BCE (see here). This dating might be somewhat bloated, possibly due to what's known as the reservoir effect, because the Nalchik archeological site is generally carbon dated to 4840–4820 BCE. However, even with the younger dating, this would still mean that early Anatolian farmer-related ancestry arrived in the North Caucasus, and thus in Eastern Europe, around 4,800 BCE at the latest. That's surprisingly early, and just too early to be relevant to any sort of Indo-European expansion from a necessarily even earlier Proto-Indo-Anatolian homeland somewhere south of the Caucasus. This means that NL122 effectively debunks Iosif Lazaridis' Indo-Anatolian hypothesis. Unless, that is, Iosif can provide evidence for a more convoluted scenario, in which there are at least two early Anatolian farmer-related expansions into Eastern Europe via the Caucasus, and the expansion relevant to the arrival of Indo-European speech came well after 5,000 BCE. I haven't done any detailed analyses of NL122 with formal stats and qpAdm. But my G25/Vahaduo runs suggest that it might be possible to model the ancestry of the Yamnaya people with around 10% admixture from a population similar to NL122.
Target: Russia_Samara_EBA_Yamnaya Distance: 3.4123% / 0.03412328 72.6 Russia_Progress_Eneolithic 18.2 Ukraine_N 9.2 Nalchik_EneolithicHowever, I don't subscribe to the idea that the Yamnaya people carry early Anatolian farmer-related admixture that spread into Eastern Europe via the Caucasus (on top of what is already found in Progress Eneolithic). Based on basic logic and a wide range of my own analyses, I believe that they acquired this type of ancestry from early European farmers, probably associated with the Trypillia culture. For instance...
Target: Russia_Samara_EBA_Yamnaya Distance: 3.2481% / 0.03248061 80.2 Russia_Progress_Eneolithic 13.6 Ukraine_Neolithic 6.2 Ukraine_VertebaCave_MLTrypillia 0.0 Nalchik_EneolithicAnother way to show this is with a Principal Component Analysis (PCA) that highlights a Yamnaya cline made up of the Yamnaya, Steppe Eneolithic and Ukraine Neolithic samples. As you can see, dear reader, there's no special relationship between the Yamnaya cline and Nalchik_Eneolithic. The Yamnaya samples, which are sitting near the eastern end of the Yamnaya cline, instead seem to show a subtle shift towards the Trypillian farmers.

Saturday, January 13, 2024
Romans and Slavs in the Balkans (Olalde et al. 2023)
It's always amusing to see some random Jovan or Dimitar arguing online that Slavic speakers have been in the Balkans since at least the Neolithic.
Obviously, Slavic peoples only turned up in the Balkans during the early Middle Ages. It's just that their linguistic and genetic impact on the region was so profound that it may seem like they've been there forever.
A new paper at Cell by Olalde et al. makes this point well. See here.
That's not to say, however, that it's an ideal effort. The paper's qpAdm mixture models probably could've been more precise and realistic. Genes of the Ancients has a useful discussion on the topic here.
Interestingly, Olalde et al. admit that they can't detect much, if any, admixture from the Italian Peninsula in the Balkans, even in samples dating to the Roman period. And yet, this doesn't stop them from accepting that the Roman Empire had a massive cultural and demographic impact on the Balkans.
I also assume that, by extension, they don't deny that Latin was introduced into the Balkans from the Italian Peninsula.
That is, Latin spread into the Balkans without any noticeable genetic tracer dye, and it eventually gave rise to modern Romanian spoken by millions of people today in the eastern Balkans. This might be a useful data point to keep in mind when discussing the spread of Indo-European languages into Anatolia.
See also...
Dear Iosif, about that ~2%
Wednesday, December 20, 2023
Dear Harald #2
The ancIBD method paper from the David Reich Lab was just published in Nature (open access here). It's a very useful effort, but the authors are still somewhat confused about the origin of the Corded Ware culture (CWC) population. From the paper (emphasis is mine):
This direct evidence that most Corded Ware ancestry must have genealogical links to people associated with Yamnaya culture spanning on the order of at most a few hundred years is inconsistent with the hypothesis that the Steppe-like ancestry in the Corded Ware primarily reflects an origin in as-of-now unsampled cultures genetically similar to the Yamnaya but related to them only a millennium earlier.This is basically a straw man argument, because it's easy to debunk. So why put it in the paper? Well, as far as I can see, to make the idea that the CWC is derived from Yamnaya look more plausible. This idea, that the CWC is an offshoot of Yamnaya, seems to be the favorite explanation for the appearance of the CWC among the scientists at the David Reich Lab. However, I'd say they're facing a major problem with that, because the CWC and Yamnaya populations have largely different paternal origins. That is, CWC males mostly belong to Y-haplogroups R1a-M417 and R1b-L51, while Yamnaya males almost exclusively belong to Y-haplogroup R1b-Z2103. Indeed, as far as I know, there are no reliable instances of R1a-M417 or R1b-L51 in any published or yet to be published Yamnaya samples. But it is possible to reconcile the Y-haplogroup data with the ancIBD results if we assume that the peoples associated with the Corded Ware, Yamnaya and also Afanasievo cultures expanded from a genetically more diverse ancestral gene pool, each taking a specific subset of the variation with them. This gene pool would've existed somewhere in Eastern Europe, probably at the western end of the Pontic-Caspian steppe, at most a few hundred years before the appearance of the earliest CWC burials in what is now Poland. Moreover, the split between the CWC and Yamnaya populations need not have been a clean one, with long-range contacts and largely female-mediated mixing maintained for generations, adding to the already close genealogical links between them. Citation... Ringbauer, H., Huang, Y., Akbari, A. et al. Accurate detection of identity-by-descent segments in human ancient DNA. Nat Genet (2023). https://doi.org/10.1038/s41588-023-01582-w See also... On the origin of the Corded Ware people
Sunday, November 19, 2023
Musaeum Scythia on the Seima-Turbino Phenomenon
A few weeks ago bioRxiv published two preprints on the Seima-Turbino Phenomenon (see here and here).
I can't say much about these manuscripts until I see the relevant ancient DNA samples, and that might take some time.
However, for now, I will say that both preprints really need to emphasize the profound impact that the Sintashta-related early Indo-Iranian speakers had on the Seima-Turbino Phenomenon. This, of course, would require Wolfgang Haak and friends to pull their heads out of their behinds and admit that the proto-Indo-Iranian homeland was in Eastern Europe, not in Iran.
At the same time, it's likely that the Seima-Turbino Phenomenon originated deep in Siberia, and its inception was probably most closely associated with the West Siberian Hunter-Gatherer (WSHG) genetic component. It's important that the preprints emphasize this too.
Moreover, I can't see any convincing arguments in either preprint that the Seima-Turbino Phenomenon was mainly associated with proto-Uralic speakers, or even that it was an important vector for the spread of proto-Uralic. So there's not much point in forcing the Uralic angle on studies focused on the Seima-Turbino Phenomenon. Indeed, what we also need is an archaeogenetics paper dealing specifically with the proto-Uralic expansion.
Apart from that, I'd like to direct your attention to the fact that Musaeum Scythia has already written a fine blog post about these preprints:
Genomic insights into the Seima-Turbino Phenomenon
See also...
Finally, a proto-Uralic genome
The Uralic cline with kra001 - no projection this time
Slavs have little, if any, Scytho-Sarmatian ancestry
Friday, November 10, 2023
Wielbark Goths were overwhelmingly of Scandinavian origin
When used properly, Principal Component Analysis (PCA) is an extraordinarily powerful tool and one of the best ways to study fine-scale genetic substructures within Europe.
The PCA plot below is based on Global25 data and focuses on the genetic relationship between Wielbark Goths and Medieval Poles, including from the Viking Age, in the context of present-day European genetic variation.
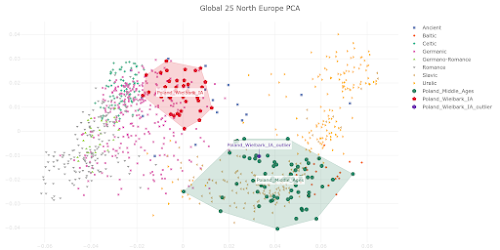 I'd say that it's a wonderfully self-explanatory plot, but here are some key observations:
I'd say that it's a wonderfully self-explanatory plot, but here are some key observations:
 Polish groups from the Middle Ages are marked with the MA suffix, while the Iron Age Wielbark Goths are marked with the IA suffix.
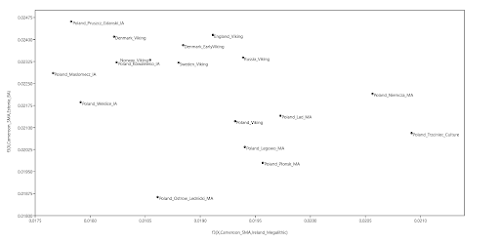
If you're wondering why I plotted the f3-statistics that I did, take a look at this (all groups largely of Scandinavian origin are emboldened):
Polish groups from the Middle Ages are marked with the MA suffix, while the Iron Age Wielbark Goths are marked with the IA suffix.
If you're wondering why I plotted the f3-statistics that I did, take a look at this (all groups largely of Scandinavian origin are emboldened):

- the Wielbark Goths (Poland_Wielbark_IA) and Medieval Poles (Poland_Middle_Ages) are two distinct populations - moreover, the Wielbark Goths form a relatively compact Scandinavian-related cluster and must surely represent a homogenous population overwhelmingly of Scandinavian origin - on the other hand, the Medieval Poles form a more extensive and heterogeneous cluster that overlaps with present-day groups all the way from Central Europe to the East Baltic, and that's because they are likely to be in large part of mixed origin - I know for a fact that at least some of these early Poles harbor recent admixture, because their burials are similar to those of Vikings and their haplotypes have been shown to be partly of Scandinavian origin (see here) - one of the Wielbark females is an obvious genetic outlier (Poland_Wielbark_IA_outlier), and basically looks like a first generation mixture between a Goth and a Balt.Please note that the PCA is only based on relatively high quality genomes, so as not to confuse the picture with spurious results and noise. Also, all outliers with potentially significant ancestry from outside of Central, Eastern and Northern Europe were removed from the analysis. The relevant datasheet is available here. However, sanity checks are always important when studying complex topics like fine-scale genetic ancestry. To that end I've prepared a graph based on f3-statistics of the form f3(X,Cameroon_SMA,Estonia_BA)/(X,Cameroon_SMA,Ireland_Megalithic), that reproduces the key features of my PCA. The relevant datasheet is available here.

f3(X,Estonia_BA,Cameroon_SMA) Poland_Legowo_MA 0.226406 Poland_Ostrow_Lednicki_MA 0.225996 Poland_Plonsk_MA 0.225017 Poland_Trzciniec_Culture 0.224215 Poland_Lad_MA 0.224142 Poland_Viking 0.223838 Poland_Niemcza_MA 0.223659 Poland_Weklice_IA 0.223549 Poland_Kowalewko_IA 0.222584 Poland_Pruszcz_Gdanski_IA 0.222324 Sweden_Viking 0.222091 Russia_Viking 0.222042 Poland_Maslomecz_IA 0.221914 Norway_Viking 0.221825 Denmark_EarlyViking 0.221257 Denmark_Viking 0.221174 England_Viking 0.220979 f3(X,Ireland_Megalithic,Cameroon_SMA) Poland_Maslomecz_IA 0.219816 Poland_Weklice_IA 0.219501 Denmark_Viking 0.2192 Poland_Kowalewko_IA 0.219176 Poland_Ostrow_Lednicki_MA 0.218916 Norway_Viking 0.218854 Poland_Pruszcz_Gdanski_IA 0.218684 Sweden_Viking 0.218626 Denmark_EarlyViking 0.218529 England_Viking 0.218308 Russia_Viking 0.217999 Poland_Viking 0.217914 Poland_Plonsk_MA 0.217756 Poland_Lad_MA 0.217719 Poland_Legowo_MA 0.21765 Poland_Niemcza_MA 0.217001 Poland_Trzciniec_Culture 0.216551Interestingly, the Middle Bronze Age samples associated with the Trzciniec Culture (Poland_Trzciniec_Culture) show a closer genetic relationship to Medieval Poles than to Wielbark Goths or Northwestern Europeans. This is indeed the case both in terms of genome-wide and uniparental markers, including some very specific lineages under Y-chromosome haplogroup R1a. But that's a much more complex issue that I'll leave for another time. So please stay tuned. See also... Slavs have little, if any, Scytho-Sarmatian ancestry
Saturday, November 4, 2023
Slavs have little, if any, Scytho-Sarmatian ancestry
Here's an abstract of a new study from the David Reich Lab about ancient Slavs, titled "Genetic identification of Slavs in Migration Period Europe using an IBD sharing graph". Emphasis is mine:
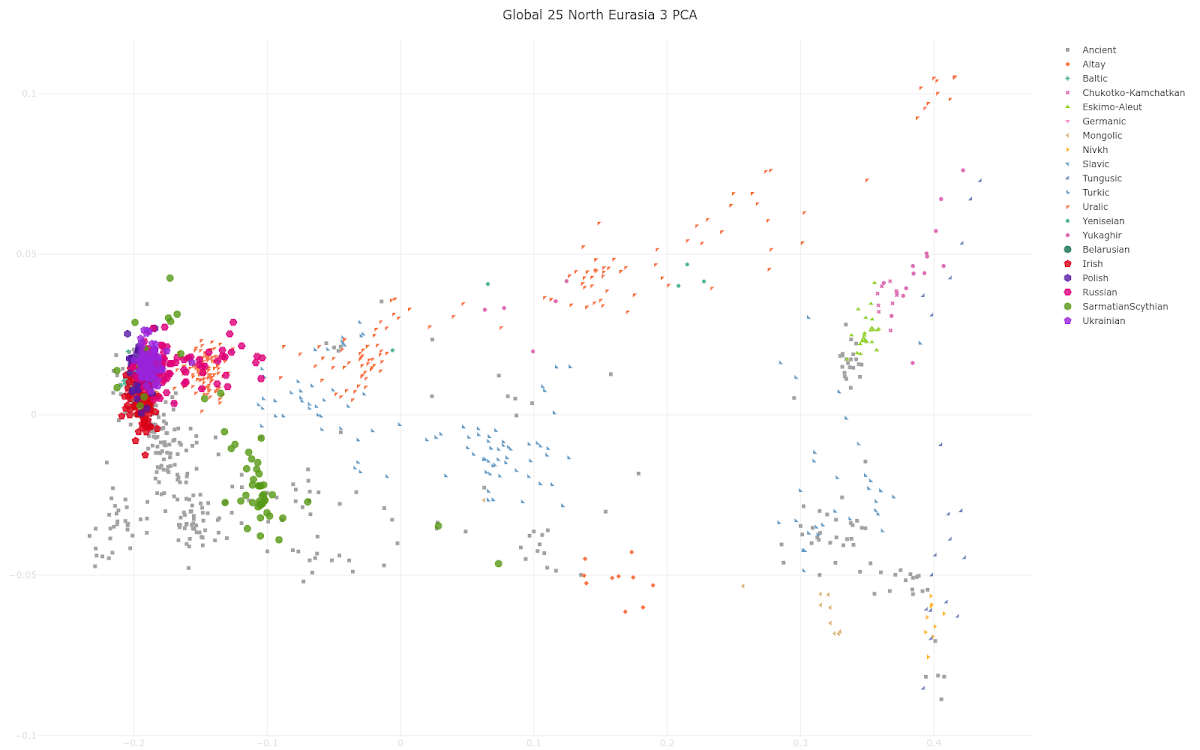 As you can see, dear reader, most of the Slavs (Belarusians, Poles, Ukrainians and many Russians) cluster with the Irish near the western end of the plot.
Some Russians are shifted significantly east of them along the "Uralic cline" and, as a result, they cluster with various Uralic speakers such as Mordovians. That's because when Slavs migrated deep into what is now northern Russia they mixed with Uralic speakers who were there before them.
Most of the Sarmatians and Scythians form a cluster southeast of the Slavs and Irish because they carry significant levels of East Asian ancestry. This type of eastern ancestry is basically missing in modern-day Slavs (see here).
Several of the Scythians cluster among the Slavs and Irish, but that's because they're genetic outliers, whose existence, if anything, suggests that some Scythians had significant Slavic-related and/or Irish-related ancestry.
Now, even though most of the Slavs do cluster with the Irish in the above PCA plot, I strongly disagree with the authors of the abstract when they claim that "differentiating Slavic, Germanic, and Celtic people is very difficult" with PCA. It's actually pretty damn easy and I've been doing it successfully for many years. For instance, see here.
See also...
Wielbark Goths were overwhelmingly of Scandinavian origin
The Caucasus is a semipermeable barrier to gene flow
As you can see, dear reader, most of the Slavs (Belarusians, Poles, Ukrainians and many Russians) cluster with the Irish near the western end of the plot.
Some Russians are shifted significantly east of them along the "Uralic cline" and, as a result, they cluster with various Uralic speakers such as Mordovians. That's because when Slavs migrated deep into what is now northern Russia they mixed with Uralic speakers who were there before them.
Most of the Sarmatians and Scythians form a cluster southeast of the Slavs and Irish because they carry significant levels of East Asian ancestry. This type of eastern ancestry is basically missing in modern-day Slavs (see here).
Several of the Scythians cluster among the Slavs and Irish, but that's because they're genetic outliers, whose existence, if anything, suggests that some Scythians had significant Slavic-related and/or Irish-related ancestry.
Now, even though most of the Slavs do cluster with the Irish in the above PCA plot, I strongly disagree with the authors of the abstract when they claim that "differentiating Slavic, Germanic, and Celtic people is very difficult" with PCA. It's actually pretty damn easy and I've been doing it successfully for many years. For instance, see here.
See also...
Wielbark Goths were overwhelmingly of Scandinavian origin
The Caucasus is a semipermeable barrier to gene flow
Popular methods of genetic analysis relying on allele frequencies such as PCA, ADMIXTURE and qpAdm are not suitable for distinguishing many populations that were important historical actors in the Migration Period Europe. For instance, differentiating Slavic, Germanic, and Celtic people is very difficult relying on these methods, but very helpful for archaeologists given a large proportion of graves with no inventory and frequent adoption of a different culture. To overcome these problems, we applied a method based on autosomal haplotypes. Imputation of missing genotypes and phasing was performed according to a protocol by Rubinacci et al. (2021), and IBD inference was done for ancient Eurasian individuals with data available at >600,000 1240K sites. IBD links for a subset of these individuals were represented as a graph, visualized with a force-directed layout algorithm, and clusters in this graph are inferred with the Leiden algorithm. One of the clusters in the IBD graph emerged that includes nearly all individuals in the dataset annotated archaeologically as “Slavic”. According to PCA a hypothesis for the origin of this population can be proposed: it was formed by admixture of a Baltic-related group with East Germanic people and Sarmatians or Scythians. The individuals belonging to the “Slavic” IBD sharing cluster form a chronological gradient on the PCA plot, with the earliest samples close to the Baltic LBA/EIA group. Later “Slavic” individuals are shifted to the right, closer to Central and Southern Europeans and probably reflecting further admixture of Slavs with local populations during the Migration Period.Apparently this abstract is causing a bit of confusion online because of the mention of possible Sarmatian or Scythian ancestry in Slavs. However, it's important to understand that the authors are referring to certain Slavic or even just Slavic-related individuals, usually from culturally heterogeneous frontier settlements deep in what is now Russia. So yes, it's possible that some of these individuals carry Sarmatian, Scythian or other exotic eastern ancestry. But even if this is true, then obviously we can't extend this inference to all ancient and modern-day Slavs. Indeed, below is a G25/Vahaduo Principal Component Analysis (PCA) that shows why modern-day Slavic speakers can't be linked genetically to Sarmatians or Scythians. To experience a more detailed version of the PCA paste the data here into the relevant field here.

Friday, September 22, 2023
The Caucasus is a semipermeable barrier to gene flow
The scientists at the David Reich Lab are a clever bunch. But they're not always on top of things. And this can be a problem.
For instance, they fail to understand that the Caucasus has effectively stymied human gene flow between Eastern Europe and West Asia through the ages. That is, the Caucasus is a semipermeable barrier to human gene flow.
 Until they accept and understand this fact, they won't be able to accurately characterize the ancestry of the ancient human populations of the Pontic-Caspian (PC) steppe, including the Yamnaya people.
In turn, they also won't be able to locate the Indo-Anatolian homeland.
Now, the Caucasus isn't a barrier to gene flow because it's difficult to cross, and, indeed, many human populations have managed to cross it since the Upper Palaeolithic. As a result, the peoples of the North Caucasus are today genetically more similar to the populations of the Near East than Europe.
In fact, the clear genetic gap between most West Asian and Eastern European populations through the ages is actually caused by the extreme differentiation between the mountain ecology of the Caucasus and the steppe ecology of the PC steppe.
That is, the Caucasus is ecologically so different from the PC steppe that it has been practically impossible for human populations to make the transition from one to the other.
Indeed, it's important to understand that there's no reliable record of any prehistoric human population successfully making the transition from the mountain ecology to the steppe ecology in this part of the world.
In other words, contrary to claims by people like David Reich and David Anthony, there's no solid evidence of any significant prehistoric human migration from the Caucasus, or from south of the Caucasus, to Eastern Europe by hunter-gatherers, farmers or pastoralists.
But, you might ask, how on earth did the Yamnaya people get their significant Caucasus/Iranian-related admixture if not via a mass migration from the Caucasus and/or the Iranian Plateau, as is often argued by the above mentioned scholars and their many colleagues?
Well, obviously, the diffusion of alleles from one population to another can happen without migration. All that is needed is a contact zone between them.
The ancient DNA and archeological data currently available from the Caucasus and the PC steppe suggest to me that there was at least one such contact zone in this area bringing together the peoples of the mountain and steppe ecoregions. This allowed them to mix, probably gradually and over a long period of time, by and large without leaving their ecoregions.
Once the Caucasus alleles entered the steppe, they were spread around by local hunter-gatherers and pastoralists who were highly mobile and well adapted to the steppe ecology.
Someone should write a paper about this.
See also...
The Nalchik surprise
Understanding the Eneolithic steppe
Matters of geography
Until they accept and understand this fact, they won't be able to accurately characterize the ancestry of the ancient human populations of the Pontic-Caspian (PC) steppe, including the Yamnaya people.
In turn, they also won't be able to locate the Indo-Anatolian homeland.
Now, the Caucasus isn't a barrier to gene flow because it's difficult to cross, and, indeed, many human populations have managed to cross it since the Upper Palaeolithic. As a result, the peoples of the North Caucasus are today genetically more similar to the populations of the Near East than Europe.
In fact, the clear genetic gap between most West Asian and Eastern European populations through the ages is actually caused by the extreme differentiation between the mountain ecology of the Caucasus and the steppe ecology of the PC steppe.
That is, the Caucasus is ecologically so different from the PC steppe that it has been practically impossible for human populations to make the transition from one to the other.
Indeed, it's important to understand that there's no reliable record of any prehistoric human population successfully making the transition from the mountain ecology to the steppe ecology in this part of the world.
In other words, contrary to claims by people like David Reich and David Anthony, there's no solid evidence of any significant prehistoric human migration from the Caucasus, or from south of the Caucasus, to Eastern Europe by hunter-gatherers, farmers or pastoralists.
But, you might ask, how on earth did the Yamnaya people get their significant Caucasus/Iranian-related admixture if not via a mass migration from the Caucasus and/or the Iranian Plateau, as is often argued by the above mentioned scholars and their many colleagues?
Well, obviously, the diffusion of alleles from one population to another can happen without migration. All that is needed is a contact zone between them.
The ancient DNA and archeological data currently available from the Caucasus and the PC steppe suggest to me that there was at least one such contact zone in this area bringing together the peoples of the mountain and steppe ecoregions. This allowed them to mix, probably gradually and over a long period of time, by and large without leaving their ecoregions.
Once the Caucasus alleles entered the steppe, they were spread around by local hunter-gatherers and pastoralists who were highly mobile and well adapted to the steppe ecology.
Someone should write a paper about this.
See also...
The Nalchik surprise
Understanding the Eneolithic steppe
Matters of geography
Subscribe to:
Posts (Atom)